Anterior Scleritis
Author: Helen Flohr
Abstract
Scleritis is a group of rare inflammatory conditions in which the sclera becomes oedematous and tender. It is often extremely painful, can be sight-threatening and is strongly associated with life-threatening systemic disease. Accurate diagnosis and treatment are important to prevent ocular and systemic morbidity. This article presents a case of diffuse, anterior, non-infectious scleritis. This form has the least degree of inflammation and is the most readily responsive to treatment with non-steroidal anti-inflammatories (NSAIDs). Treatment of more severe forms of scleritis with greater degrees of inflammation can require oral corticosteroids or immunomodulatory agents.
Introduction
Scleritis is a group of rare inflammatory conditions in which the sclera becomes oedematous and tender. It can be recurrent. It ranges from a benign, self-limiting inflammation to a severe form with necrosis of the underlying sclera. It is classified thus, from most to least common:

- Anterior scleritis
- Non-necrotizing
- Diffuse
- Nodular
- Necrotizing
- With inflammation
- Without inflammation (scleromalacia perforans)
- Posterior scleritis 1
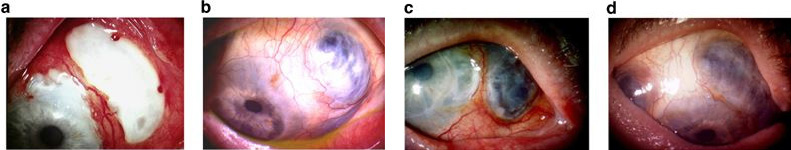
Scleritis can be unilateral or bilateral and onset is usually gradual over several days1. It is characterised by severe and boring eye pain which may radiate to the forehead, brow or jaw and may awaken the patient at night. Other signs include red eye, tearing, photophobia and reduced vision if there is concurrent inflammation of the cornea or uveal tract2. Anterior scleritis is characterised by oedema of the sclera and dilation of both the superficial and deep episcleral vasculature. This results in a bluish hue to the involved sclera, best seen under natural light. Anterior, non-necrotizing scleritis is the most common form and is characterised as either diffuse, which can involve the entire anterior sclera or just a segment, or nodular, in which there is also a visible, immovable elevation with engorged scleral vessels 3. Anterior necrotizing scleritis with inflammation is a severe form of the disease. It is frequently associated with life-threatening vasculitis and carries a poor prognosis1. Vascular occlusion results in blanched, avascular patches of sclera, followed by scleral thinning and exposure of the underlying uveal tissue. In both non-necrotizing and necrotizing forms, inflammation can spread to the surrounding structures, resulting in keratitis, uveitis, glaucoma, cataract or exudative retinal detachment2. Ocular complications and vision loss are significantly more common with necrotizing and posterior scleritis3,4.
Aetiology and Associations
The major inflammatory cells found in the deep episcleral tissue during scleritis are T cells and macrophages, with clusters of B cells in perivascular areas. HLA-DR expression and increased interleukin-2 (IL-2) expression on T cells has been documented, suggesting an active, cell-mediated immune response5. T-helper lymphocytes (Th17 cell) have been implicated in the pathogenesis of human autoimmune diseases, including non-infectious scleritis. These cells can be promoted by interleukin-2 (IL-2) or inhibited by interferon gamma (IFN-?)6. Therapeutic agents that target these chemokines may be of benefit in treatment of recalcitrant cases unresponsive to primary treatment methods.
Anterior scleritis is associated with autoimmune disease in 36-57% of cases, depending on the clinical setting4,3, but can also be infectious (5% of cases) or idiopathic. The most common systemic associations are the connective tissue diseases (rheumatoid arthritis, Wegener’s granulomatosis, inflammatory bowel disease, systemic lupus erythematosus and relapsing polychondritis). One study found scleritis to be the first manifestation of a connective tissue or vasculitic disease in 38.7% of patients; diffuse anterior scleritis was found to be the most frequent type of scleritis in these patients4. Infectious causes, including post-herpes zoster ophthalmicus, syphilis, tuberculosis and Lyme disease7, are less common but have a higher incidence of ocular morbidity, with only 33-50% of eyes retaining vision better than 6/60 after treatment, and up to 25% of eyes requiring enucleation8.
Case Report

A 53 year old male, LB, was referred by his general practitioner with a two week history of a red, mildly photophobic right eye and a deep, boring pain behind the eye. He reported very good general health and was on no medications. Specifically, there was no history of lower back pain, arthritis or other autoimmune disease. He had no peptic ulcer disease or gastro-oesophageal reflux disease. He had a history of metal corneal foreign bodies, but otherwise had not experienced any other ocular inflammatory episodes.
Visual acuity was 6/4.5 in each eye. Pupil reactions were normal with no relative afferent pupil defect and motility was full. Dilated fundus examination was unremarkable.
Slit lamp examination revealed moderate, diffuse injection of both the superficial and deep episcleral vessels in a supero-temporal sector of the sclera of the right eye. The deep episcleral vessels did not bleach in response to 2.5% phenylephrine. Palpation of the inflamed area through the eyelid was acutely painful. A mild acute anterior uveitis was present, with trace cells in the anterior chamber and fine keratic precipitates (KP) on the endothelium. The left eye was quiet. Intraocular pressures were R 7mm Hg, L 16mm Hg.
LB was diagnosed with diffuse anterior scleritis with associated anterior uveitis. He was prescribed ibuprofen 400mg qid to be taken with food for the scleritis. The anterior uveitis was treated with Prednefrin forte q2h for two days then qid, and homatropine 2% tid. The association of scleritis with systemic autoimmune disease was addressed with the patient, and he was referred to his general practitioner for further investigations.
At review five days later, LB reported that although he had experienced some ocular pain with heavy lifting two days earlier, the eye was now comfortable. He experienced no gastric discomfort with the ibuprofen. Slit lamp examination showed mild, diffuse injection of the involved sector of sclera and there was no pain on palpation. The anterior chamber was quiet, with no cells, and just a few KP on the corneal endothelium. Intraocular pressures were R11, L 14.
LB was advised to stop the homatropine, to taper the Prednefrin Forte to tid, and to continue the ibuprofen at the same dose of 400mg qid until the next review in four days. At this subsequent review, the patient was very comfortable, with a white eye, quiet anterior chamber and no KP on the endothelium. He was advised to cease the ibuprofen and to taper the Prednefrin Forte gradually over the next eight days. The patient was reminded of the possibility of recurrence and to see his general practitioner for investigation of potential autoimmune associations. At the time of writing, no information had been received regarding these investigations.
Differential Diagnosis
Scleritis should be distinguished from episcleritis. Episcleritis has a more acute onset and involves only the superficial episcleral vessels which can be bleached with phenylephrine 2.5%2, while the deep episcleral vessels that are involved in scleritis do not bleach. In scleritis, the involved sclera often has a bluish hue, best seen in natural light, while the inflamed area in episcleritis has a more salmon pink colour. Perhaps most tellingly, scleritis also involves severe pain, and palpation of the affected area elicits an extreme response, whereas episcleritis tends to only cause mild tenderness.
Ocular complications occur more often in scleritis than episcleritis. These include keratitis, uveitis, and glaucoma with anterior disease; and exudative detachments or other posterior segment complications with posterior scleritis. When anterior scleritis is seen, fundus examination is essential to check for additional posterior involvement and inflammation of the retina, choroid and optic nerve head. Proptosis and limitation of movement of the extraocular muscles suggest involvement of the posterior sclera1.
Therapeutic Management
Treatment of non-infectious, non-necrotizing scleritis involves a stepped regime of oral NSAIDs, followed by oral steroids and finally steroid sparing agents. One study found that of all scleritis patients, 30.4% required oral NSAIDs, 31.9% oral prednisone and 26.1% systemic immunosuppressive drugs. Patients with idiopathic diffuse or nodular scleritis with a low degree of scleral inflammation, or without ocular complications are most likely to respond to NSAIDs9. Those with necrotizing scleritis and posterior scleritis are more likely to require oral corticosteroids or immunosuppressive drugs (90% and 100% respectively) than are patients with diffuse anterior and nodular anterior scleritis (56.4% and 21.4% respectively)3. Decreased pain indicates a response to treatment, even if the inflammation appears unchanged2.
Oral NSAIDs are considered first-line therapy for scleritis for their ease of use, cost and relatively mild side-effect profile. The most commonly used NSAIDs include indomethacin, naproxen and ibuprofen, as was seen to be effective in this case. Short-term use is often well-tolerated, but can be associated with peptic ulcer disease, hypertension, heart disease, bleeding, fluid retention, renal disease and mood change and should be used with caution in those patients with increased risk of these conditions2.
Systemic steroids are highly recommended for the treatment of severe or necrotizing scleritis, or when oral NSAIDS have not been effective within a week1. Oral prednisone is typically used, usually beginning at a dose of 1mg/kg/day, for several days, with a slow taper over several weeks5.
If there is minimal response to oral prednisone after a month, additional agents which target the inciting inflammatory cell may be required. Immunomodulatory therapies include antimetabolites (eg methotrexate), alkylating agents (chlorambucil and cyclophosphamide) and T-cell inhibitors (cyclosporine and tacrolimus). Second-line biologic therapies which reduce proliferation of T and B cells include anti-TNF agents such as etanercept, or monoclonal antibodies which target the IL-2 receptor on T cells. Research into other biologic therapies which target specific cytokines is ongoing 6, 8.
Topical steroids are useful for treating coexisting intraocular inflammation. Prednefrin Forte is often used for mild cases of anterior scleritis associated with intraocular inflammation, such as the low-grade uveitis seen in this case. Scleritis does not typically respond to topical corticosteroids alone3 and oral agents are usually required, however single injections of subconjunctival or sub-Tenon’s triamcinolone have been found to be effective and safe in cases of non-necrotizing, non-infectious scleritis. It should not be used in necrotizing scleritis due to increased risk of progressive scleral necrosis and perforation8.
As scleritis is frequently a manifestation of systemic disease, it is important to exclude multi-system disease at presentation. Every patient should undergo tests including complete blood count, erythrocyte sedimentation rate (ESR), uric acid, rapid plasma reagin (RPR), FTA-ABS, rheumatoid factor (RF), antinuclear antibody (ANA) and fasting blood sugar. Serum anti-neutrophilic cytoplasmic antibody (c-ANCA) can be obtained if Wegener’s granulomatosis is suspected. Other tests if warranted include PPD with anergy panel, chest x-ray, x-ray of sacroiliac joints, B-scan ultrasonography to detect posterior scleritis, MRI or CT scan1, 2.
Conclusion
Scleritis is a severe ocular inflammation, often associated with ocular complications and systemic autoimmune disease. The treatment paradigm for non-infectious, non-necrotizing scleritis is a stepped approach involving oral NSAIDs, followed by oral steroids and finally steroid sparing agents, along with appropriate detection and management of coexisting ocular and systemic inflammation. Diffuse anterior scleritis is the most common form of scleritis, generally has the lowest degree of inflammation and can often be managed with oral NSAIDs alone.
References
- Watson PG, Hayreh SS. Scleritis and episcleritis. Brit J Ophthal. 1976; 60: p. 163-191.
- Cullom RD Jr, Chang B. The Wills Eye Manual Office and Emergency Room Diagnosis and Treatment of Disease. Second Edition ed. Philadelphia: J B Lippincott Company; 1994.
Jabs DA, Mudun A, Dunn JP, Marsh MJ. Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol. 2000; 130: p. 469-476.
- Sainz de la Maza M, Molina N, Gonzalez-Gonzales LA, Doctor PP, Tauber J, Foster S. Clinical characteristics of a large cohort of patients with scleritis and episcleritis. Ophthalmology. 2012; 119: p. 43-50.
- Okhravi N, Odufuwa B, McCluskey P, Lightman S. Scleritis. Surv Ophthalmol. 2005; 50: p. 351-363.
- Rachitskaya A, Mandelcorn ED, Albini TA. An update on the cause and treatment of scleritis. Curr Opin Ophthalmol. 2010; 21: p. 463-467.
- Pavesio CE, Meier FM. Systemic disorders associated with episcleritis and scleritis. Curr Opin Ophthalmol. 2001; 12: p. 471-478.
- Beardsley RM, Suhler EB, Rosenbaum JT, Lin P. Pharmacotherapy of scleritis: current paradigms and future directions. Expert Opin. Pharmacother. 2013; 14: p. 411-424.
- Sainz de la Maza M, Molina N, Gonzalez-Gonzalez LA, Doctor PP, Tauber J, Foster S. Scleritis Therapy. Ophthalmology. 2012; 119: p. 51-58.
- Bruce AS, Loughnan MS. Anterior Eye Disease and Therapeutics A-Z London: Butterworth-Heinemann; 2003.
Practice Location
Logan City Centre
Cnr Wembley and Kingston Roads
Logan Central, Qld 4114
Ph: 07 3299 3699
Fax: 07 3290 4558
Email: via our contact page
Operating Hours
Monday 8:30am - 5:00pm
Tuesday 8:30am - 5:00pm
Wednesday 8:30am - 5:00pm
Thursday 8:30am - 7:00pm
Friday 8:30am - 5:00pm
Saturday 8:30am - 12:00pm
Sunday Closed